それぞれの原子に異なる基底関数を指定する
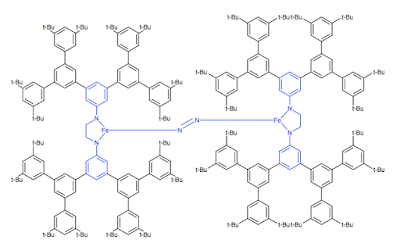
非常に大きな分子の計算において、末端置換基を切断してしまい、計算を軽くする方法がよく知られています。 徐々に基底関数を高級なものにしていく手法 と並行し、かぼそい計算パワーしか使うことのできなかった時代の知恵です。 もちろん、知りたいことがその計算でわかればよいのですが、嵩高さゆえの歪んだ構造を計算したい場合、まったく役に立ちません。やっぱり、切り捨てちゃだめだよね。。。 例えば、こんな分子(架空のものです)。青い部分をマジメに計算すれば、 電子状態はほとんど記述できるでしょう。 しかし外側をバッサリ切ると、中心構造にも影響が。 ONIOMの設定が、現在gauss viewでは手軽にできるようなので、それを勉強するのは解決法の一つかと思います(私はまだつかえませんが)。 オルタナティブな方法として、各々の原子に基底関数を割り当てられることを利用して、中心の電子状態を決定している部分には、大きな基底関数を当て、末端の嵩高さだけの寄与(だとかんがえている)の部分に小さな基底を当てる手法を紹介します。 gen キーワードを使うと元素毎に基底関数を当てられること は紹介しましたが、それの応用版です。 原子ごとに番号が与えられていますが、これを利用します。 原子の番号は、gauss viewをつかっていれば、下の絵のコマンドで簡単に確認できるので参考にしてください。 あるいは、gauss view 上で、原子の上にマウスカーソルを持ってくると、左下に表示がでます。 ここから_______ %nprocshared=16 %mem=63000MB %chk=Fe.chk # freq b3lyp/gen geom=allcheck guess=read int=ultrafine Title card required 2 5 Fe 1.59676500 -0.03311300 0.00000000 N -0.05620400 0.09399000 -1.32295000 C 0.08688200 -0.02090700 -2.73226500 C