自然軌道解析(NBO解析)による錯体の価数の決め方

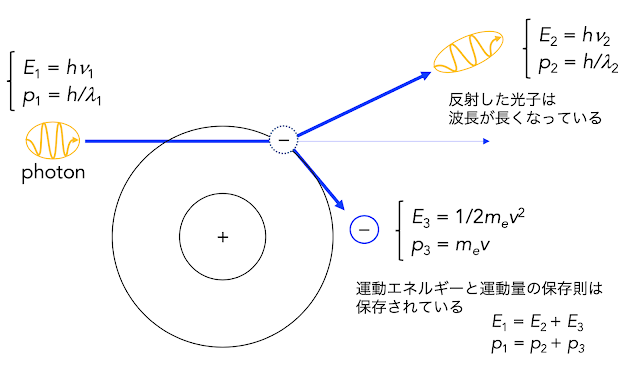
NBOを利用する手法についての解説、第二弾です。(第一弾は こちら ) 興味を持っている分子について、どんなふうに電子が分布しているか、しりたいですよね! Mulliken電子密度は、計算化学を初めて最初に触れる電子密度についての情報だと思います。Mullken電子密度は、小さな有機物について考える場合なんかは十分なパラメータなのですが、少し重たい元素が入ってくると、問題になることも多いそうです。 いにしえの頃、非常に原始的なSTOを 基底関数にして計算していた時にはそれで良かったようですが、diffuse関数や、分極関数が存在する基底ではまずいそうだ。 名大の計算機センターのpdf に詳しいです。 そもそも分子の中で非局在化している電子の所在は、なかなか特定しづらいものです。 (だから価数なんて考えてもしゃーないやん、とか言う人もいらっしゃいます。私とは宗派が違いますが。) ざっくりとMulliken電子密度の問題点を書くと、それぞれの原子の軌道の形などを考慮せず、結合の適当な位置にズバッと線を引いて、電子の数をカウントするところが問題です。 水素分子のような等核2原子分子ならど真ん中でOKでしょうが、C–H結合の真ん中に線をひいても、正しい価数のカウントにはなりませんよね?たとえばsp2混成の場合とsp3混成の場合で、線を引く位置は変わりそうです。 分子骨格が似た分子同士で、原子上のMuliken電子密度を比較することには使えそうですが、それ以外の用途には厳しい手法です。 私は遷移金属錯体の計算を行いますが、たとえばどっからどう見ても 2価の銅 錯体のテトラアンミン銅錯体のCu上に乗っかっている Mulliken Chargeは +0.744 で、全然 +2 からは遠い (図1)。 図2. テトラアンミン銅(II)錯体のMulliken Charge Density あれ、銅のアンモニア錯体は平面ちゃうの?と思ったあなたは鋭い。この計算では、カウンターアニオンや溶媒の影響を考慮していませんが、どうやら、軸位に相互作用するものがまったくない場合、少し四面体様の歪を持つみたいです。CCDCの構造検索も軽くして見ましたが、だいたい軸位に何かが弱く相互作用すると平面錯体になるようですね。